Drug Induced Liver Injury
Introduction
Drug metabolism in liver
The liver is considered to be the "metabolic clearing house" for both endogenous chemicals ( steroids, cholesterol, protein etc) and xenobiotics.
Drug metabolism encompasses three Phases:
Phase I reactions: Involves introduction of a functional group (-OH, -NH2, -SH or –COOH) or exposure of functional group. The phase 1 oxidation reactions are carried out by Cytochrome P450 group of enzymes (CYPs,) flavin-containing monooxygenases (FMO), and epoxide hydrolases (EH).
The various reactions in this group are as follows:
| Cytochrome P450-dependent oxidations: | |
| Aromatic hydroxylations | Acetanilide, propranolol, phenobarbital, phenytoin, phenylbutazone, amphetamine, warfarin, 17-ethinyl estradiol, naphthalene, benzpyrene |
| Aliphatic hydroxylations | Amobarbital, pentobarbital, secobarbital, chlorpropamide, ibuprofen, meprobamate, glutethimide, phenylbutazone, digitoxin |
| Epoxidation | Aldrin |
| Oxidative dealkylation | |
| N-Dealkylation | Morphine, ethylmorphine, benzphetamine, aminopyrine, caffeine, theophylline |
| O-Dealkylation | Codeine, p-nitroanisole |
| S-Dealkylation | 6-Methylthiopurine, methitural |
| N-Oxidation | |
| Primary amines | Aniline, chlorphentermine |
| Secondary amines | 2-Acetylaminofluorene, acetaminophen |
| Tertiary amines | Nicotine, methaqualone |
| S-Oxidation | Thioridazine, cimetidine, chlorpromazine |
| Deamination | Amphetamine, diazepam |
| Desulfuration | Thiopental, Parathion |
| Dechlorination | Carbon tetrachloride |
| Cytochrome P450-independent oxidations: | |
| Flavin monooxygenase (Ziegler's enzyme) | Chlorpromazine, amitriptyline, benzphetamine, Desipramine, nortriptyline, Methimazole, propylthiouracil |
| Amine oxidases | Phenylethylamine, epinephrine |
| Dehydrogenations | Ethanol |
| Reductions |
|
| Azo reductions | Prontosil, tartrazine |
| Nitro reductions | Nitrobenzene, chloramphenicol, clonazepam, dantrolene |
| Carbonyl reductions | Metyrapone, methadone, naloxone |
| Hydrolyses |
|
| Esters | Procaine, succinylcholine, aspirin, clofibrate, methylphenidate |
| Amides | Procainamide, lidocaine, indomethacin |
The phase 2 enzymes include several super families of conjugating enzymes, the more important ones being the glutathione-S-transferases (GST), UDP-glucuronosyltransferases (UGT), sulfotransferases (SULT), N-acetyltransferases (NAT), and methyltransferases (MT). Each super family of enzymes contains multiple genes which accounts for the individual variation in handling of the same drug by different people/ race.
Types of Phase 2 metabolic reactions:
| Type of Conjugation | Transferase (Location) | Types of Substrates | Examples |
| Glucuronidation | UDP glucuronosyltransferase (microsomes) | Phenols, alcohols, carboxylic acids, hydroxylamines, sulfonamides | Nitrophenol, morphine, acetaminophen, diazepam, N-hydroxydapsone, sulfathiazole, meprobamate, digitoxin, digoxin |
| Acetylation | N–Acetyltransferase (cytosol) | Amines | Sulfonamides, isoniazid, clonazepam, dapsone, mescaline |
| Glutathione conjugation | GSH-S-transferase (cytosol, microsomes) | Epoxides, arene oxides, nitro groups, hydroxylamines | Acetaminophen, ethacrynic acid, bromobenzene |
| Glycine conjugation | Acyl-CoA glycinetransferase (mitochondria) | Acyl-CoA derivatives of carboxylic acids | Salicylic acid, benzoic acid, nicotinic acid, cinnamic acid, cholic acid, deoxycholic acid |
| Sulfation | Sulfotransferase (cytosol) | Phenols, alcohols, aromatic amines | Estrone, aniline, phenol, 3-hydroxy-coumarin, acetaminophen, methyldopa |
| Methylation | Transmethylases (cytosol) | Catecholamines, phenols, amines | Dopamine, epinephrine, pyridine, histamine,thiouracil |
Incidence:
An adverse drug reaction is a “response to a drug which is noxious and unintended and which occurs at doses normally used in man for prophylaxis, diagnosis, or therapy of disease or for the modification of physiologic function.” DILI is one of the important adverse drug reactions.The annual incidence is said to be between 1 in 10,000 to 1,00,000; however, incidences as high as 14 per 100,000 population have been reported. Drug induced liver injury accounts for up to 10 percent of all adverse drug reactions. Furthermore DILI is responsible for more than 50% of all cases of acute liver failure with paracetamol being the principal offending agent.
Mechanistic Classification of drug-induced liver injury:
Type A / Intrinsic hepatotoxicity: are predictable, dose related and have a short and consistent latency period between drug exposure and liver injury. Can result from
- Direct hepatotoxins: destroy the hepatocytes by physicochemical attack.There are no known direct hepatotoxins that are used as therapeutic agents e.g CCl4
- Indirect hepatotoxins: they cause cytotoxicity either by competitive inhibition of essential metabolite/interfering with selective metabolic pathway which is essential for the cellular function e.g alcohol, acetaminophen,tetracycline,mercaptopurine (antimetabolites) Or Interfere with the secretory process of the hepatocytes resulting in cholestasis e.g anabolic steroids, OCP's.
Type B/ Idiosyncratic hepatotoxicity: these are unpredictable, non dose related, based on the individual susceptibility. Latency period may vary from, few days to 1 year. May be due to:
- Immunologic injury/ hypersensitivity reaction: accompanied by fever, rash, eosinophilia, corresponding histological findings. e.g: methyldopa,phenytoin, erythromycin, sulfonamides.
- Aberrant drug metabolism: not associated with above mentioned features. Attributable to acquired or genetic variations in the enzymes involved in drug metabolism. e.g: valproic acid hepatotoxicity due to acquired or inherent deficiency of beta oxidation resulting in the formation of toxic metabolite 4-envalproate.
It is those drugs which cause Type B reactions that escape detection during drug development. stressing the need and importance of post marketing surveillance and ADR reporting.
A General 3-Step Model for drug-induced liver injury:
DILI is not sufficiently characterized by the initial injury, but always involves several mechanisms, regulatory systems and risk factors with complex interactions. A general mechanistic model is presented in depicted in the figure below. Mitochondria stand at the center of life and death in hepatotoxicity indicating the central role of mitochondria in pathogenesis.
Step 1- Initial Mechanisms of Toxicity:
- Direct Cell Stress: Reactive metabolites can exert initial cell stress through a wide range of mechanisms including depletion of glutathione (GSH), or binding to enzymes, lipids, nucleic acids and other cell structures. Furthermore reactive metabolites or parent drugs may specifically inhibit other hepatocellular functions such as the apical (canalicular) bile salt efflux pump (BSEP) in which case the subsequent intracellular accumulation of its substrates may cause secondary toxic hepatocyte damage.
- Direct Mitochondrial Impairment: Reactive metabolites or parent drugs uncouple or inhibit the mitochondrial respiratory chain causing ATP depletion and increased concentrations of reactive oxygen species (ROS), inhibit beta oxidation leading to steatosis, damage mitochondrial DNA or interfere with its replication, or directly cause mitochondrial permeability transition (MPT), i.e. opening of the “MPT pore” located in their inner membrane. The inhibition of mitochondrial electron transport in the early stages will not be reflected by elevated ALT values, indicating the requirement for earlier markers of impending mitochondrial damage. Functional tests such as the 13C-methionine breath test or the use of NMR spectra for metabonomics may prove to be useful here.
- Specific Immune Reactions: Specific immune responses involving cytotoxic T-cells with concomitant release of inflammatory cytokines can be evoked by reactive metabolites that covalently bind to proteins and are subsequently recognized as neo-antigens (hapten formation). Their subsequent Major Histocompatibility Complex (MHC) -dependent presentation on antigen presenting cells (kupffer cells) may then activate formation of antibodies against haptens or autoantibodies against cell structures such as CYP450 enzymes. In some instances initial injury also targets non-parenchymal liver cells, biliary epithelial cells, stellate cells and the sinusoidal lining. Different hepatotoxins are typically associated with specific patterns in their initial mechanism of injury. These initial specific injurious mechanisms can also be referred to as 'upstream events'. In the next step they lead to subsequent rather unspecific 'downstream events' that involve the innate immune system, which balances pro- and anti-inflammatory responses and therefore determines the further progress to severe injury or recovery.
Step 2 - Direct and Death Receptor-Mediated Pathways Leading to Mitochondrial Permeability Transition:
If the initial mechanism does not directly target and impair mitochondrial function, step 2 occurs in two principle ways, i.e. either via a direct pathway or via an indirect death receptor-amplified pathway.
In the intrinsic pathway severe intracellular stress activates c-jun N-terminal kinase (JNK). Subsequent activation of pro-apoptotic (e.g. Ba-x, Ba-k, Ba-d) proteins and inhibition of anti-apoptotic (e.g. Bcl-2, Bcl-xL) proteins of the Bcl-2 family activates MPT. If the initial event is a specific immune reaction, MHC-dependent antigen presentation will activate the release of TNF- α and FasL from Kupffer cells (hepatic macrophages) and cytotoxic T-cells.
According to the danger hypothesis for autoimmune diseases, haptenization alone may be insufficient to trigger the development of frank allergic hepatotoxicity, which requires an additional stimulation, a so called danger signal. If reactive metabolites cause concomitant mild direct cell stress or if other concomitant inflammatory diseases are present, the accompanying release of injurious cytokines may constitute such a danger signal that promotes MHC II dependent antigen presentation which renders hepatocytes more susceptible to injury, and therefore promotes autoimmune hepatotoxicity.
Regardless of how the extrinsic pathway is initiated, eventually TNF α and FasL bind to intracellular death receptors The activating death-receptor complex is also called the death-inducing signaling complex (DISC). Caspase 8 can activate pro-apoptotic Bcl-2 proteins (e.g. Bi-d), as well as signaling ceramides. Like in the intrinsic pathway this leads to MPT, which therefore plays a key role and is a common step that mediates further cell death in both, intrinsic as well as extrinsic pathways.
Step 3 - Apoptosis and Necrosis
Impaired mitochondrial function and energy production leads to apoptotic or necrotic cell death. MPT allows massive influx of protons through the inner mitochondrial membrane, which stops mitochondrial ATP synthesis. Matrix expansion and mitochondrial outer membrane permeabilization and rupture with release of cytochrome c and other pro-apoptotic mitochondrial proteins from the intermembrane space into the cytosol then takes place. In the case of apoptosis, cytochrome C then binds to a cytoplasmic scaffold (apaf-1) and pro-caspase 9, forming a complex called apoptosome, which activates signaling procaspase 9. These processes eventually result in programmed apoptotic cell death, which is characterized by cytoplasmic and nuclear condensation and fragmentation without loss of membrane integrity.
Pattern Of Drug Induced Liver Injury:
It is important to recognize the pattern of liver injury, since certain drugs tend to create injury predominantly according to one pattern or the other. The three main patterns of DILI are:
1. Hepatocellular
2. Cholestatic
3. Mixed Others : vascular and neoplastic .
Clinical presentation:
It can present as as acute (clinical or histopathological evidence present for <3>3 mths) liver disease. Many drugs create a pattern of injury that has characteristic biochemical, clinical, histology and chronologic features, or a combination of them. Together, these features form what is termed a drug's signature disease.
- Asymptomatic elevations in aminotransferase levels :
- Acute drug induced liver disease:
LESION | SYNDROME | CLINICAL PRESENTATION | EXAMPLES |
1. Hepatocellular |
|
|
|
Necrosis | Acute hepatitis | A,N,V | INH, diclofenac, halothane |
Sinusoidal beading | Pseudomononucleosis | A,N,V,L,S | Phenytion, dapsone |
Massive | Acute liver failure | A,N,V,L,S,C,E | Phenytion, dapsone |
Subacute | Prolonged hepatitis | A,N,V,J,FG | Propylthiouracil. diclofenac |
Spotty | subclinical | NONE | INH, diclofenac |
STEATOSIS (Microvesicular) | Reye-like | N,V,E | tetracycline |
2.Cholestatic |
|
|
|
Hepatocanalicular(bile (casts, portal inflammation) | Obstructive jaundice | A,N,PR,J,FV,R,P | Erythromycin Carbamazepine sulfonylureas |
Canalicular(bile casts) | Obstructive jaundice | PR,J,P | Anabolic steroids,OCPS |
1. MIXED: most common form, have mixed features. E.g captopril, trazodone, clindamycin | |||
A, Anorexia; N,nausea;,v,vomiting; l,lymphadenopathy; s,spleenomegaly; c,coagulopathy; e,encephalopathy; j,jaundice; FG,fatigue; PR,prutitus; FV,fever; p,pain; r,rash.
- Chronic drug induced liver disease:
Chronic hepatitis — Four types of chronic hepatitis have been associated with drug-induced hepatotoxicity . recognition is important because withdrawal of offending drug may lead to marked improvement or complete resolution of injury with in 4 wks.
- Autoimmune hepatitis like presentation. There is a female preponderance, serologic markers of autoimmunity (ANA, smooth muscle antibody), hyperglobulinemia, and histological features consistent with autoimmune hepatitis . The clinical manifestations range from asymptomatic biochemical abnormalities to development of cirrhosis. E.g methyldopa, minocycline, nitrofurantoin, diclofenac, fenofibrate, papaverine, phenytoin, propylthiouracil, germander, statins
- Viral hepatitis-like syndrome . The antibodies are directed against isoforms of Cyt P450 or other microsomal cellular components . hypergammaglulinemia is not a prominent feature . E.g phenytoin, dihydralazine, and ticrynafen.
- A third rare type of injury leads to a syndrome with the histologic characteristics of chronic hepatitis, but without autoimmune serologic markers. E.g lisinopril, sulfonamides, and trazodone.
- Rarely, drugs may lead to chronic toxicity without active necroinflammatory disease. Examples include dantrolene, aspirin, and isoniazid.
Steatosis — predominantly macrovesicular, tends to be less severe than that seen with acute microvesicular steatosis. commonly manifestats as hepatomegaly. Steatosis may remain asymptomatic, or may evolve into steatohepatitis with progression to cirrhosis .Drug-induced steatohepatitis may resemble alcoholic liver disease or NASH . e.g diethylstilbestrol, glucocorticoids, griseofulvin, methotrexate, nifedipine, tamoxifen, total parenteral nutrition, mercury, and ethanol.
Fibrosis and cirrhosis — Progressive liver injury leads to scarring with subsequent cirrhosis. Drug-induced cirrhosis may result from steatosis (amiodarone) or chronic hepatitis, chronic cholestasis etc . Gradual progression to cirrhosis can be seen without any manifestation of clinical illness (as with methotrexate or methyldopa).
Phospholipidosis — Phospholipidosis occurs frequently and is caused by amphophilic drug that accumulates with in the lysosomes is rare. commonly seen after prolonged administration of the offending agent. Predominant feature is hepatomegaly and mild liver dysfunction. There appears to be a high incidence of cirrhosis associated with this lesion. E.g amiodarone, amitriptyline, chloroquine, perhexilene maleate, chlorpheniramine.
Chronic intrahepatic cholestasis - produces a syndrome resembling primary biliary cirrhosis but serum antimitochondrial antibodies are usually not seen . Most cases resolve with withdrawal of the offending agent. However, some patients develop a vanishing bile duct syndrome and liver failure . e.g amitriptyline, ampicillin, amoxicillin-clavulanate, carbamazepine, chlorpromazine.
Biliary sclerosis — commonly seen after therapy of metastatic carcinoma with intrahepatic arterial infusion of floxuridine . The lesions resemble primary sclerosing cholangitis . Affected patients present with right upper abdominal pain, anorexia, weight loss, and jaundice.
Vascular disease — Drug-induced hepatic vascular disease is uncommon but can have several presentations.
- Hepatic vein thrombosis — Hepatic venous outflow obstruction (Budd-Chiari syndrome) may arise from drug-induced thrombosis of the hepatic veins or inferior vena cava. E.g Most common drug is oral contraceptives .
- Hepatic sinusoidal obstruction syndrome (formerly known as veno-occlusive disease) - resembles the Budd-Chiari syndrome clinically but is due to injury and fibrotic occlusion of the terminal hepatic venules. E.g pyrrolizidine alkaloids (found in herbal remedies) , azathioprine, mercaptopurine, vitamin A, oral contraceptives.
- Peliosis hepatis — Peliosis hepatis is rare and characterized by multiple, small, dilated blood-filled cysts in the hepatic parenchyma. E.g anabolic steroids, azathioprine, mercaptopurine, oral contraceptives, danazol. Lesions may resolve with discontinuation of the offending agent.
Granulomatous disease — Noncaseating granulomas in the liver can be seen with a variety of drugs .presents with low-grade fever and chronic fatigue; jaundice is rare. The injury is usually transient without significant sequelae. e.g allopurinol, amiodarone, carbamazepine, diazepam, diltiazem, isoniazid, methyldopa, phenytoin, and sulfonylureas
Neoplasia
- Hepatic adenoma — The risk of hepatic adenoma is increased in women taking oral contraceptives (OCP) and in men taking anabolic steroids. Adenomas have historically developed in women using high-dose OCPs or use for greater than five years. Adenomas are now more rare as the level of estrogen in OCPs has fallen.
- Angiosarcoma — This extremely rare tumor has been associated with use of thorotrast , potassium arsenite, polyvinyl chloride, and anabolic steroids.
- Hepatocellular carcinoma — The main chemical associations with the development of hepatocellular carcinoma include aflatoxin, oral contraceptives, and alcohol.
Risk factors:
- Race: Some drugs appear to have different toxicities based on race. For example, blacks and Hispanics may be more susceptible to isoniazid (INH) toxicity. The rate of metabolism is under the control of P-450 enzymes and can vary from individual to individual.
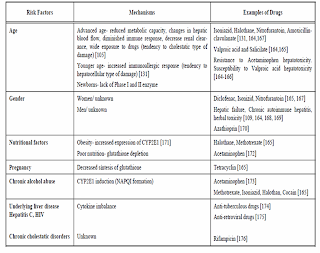
- Alcohol ingestion: Alcohol causes depletion of glutathione (hepatoprotective) stores and induce cyp2E1 that make the person more susceptible to toxicity by drugs.
- Drug formulation: Long-acting drugs may cause more injury than shorter-acting drugs.
- Drug interactions: concomitant use of drugs or herbal formulation or dietary supplements can affect the metabolism of other drugs ( enzyme induction and inhibition) resulting in accumulation of toxic meatabolites and subsequent injury.

Diagnosis:
The clinical diagnosis and prediction of DILI remains a major challenge due to wide spectrum of manifestation from asymptomatic rise of ATs to hepatic failure and of course various confounding factors( preexisting liver disease, multiple drug usage by patients, and most important, lack of reliable screening methods and diagnostic standards) make it still more complex.
Hyman Zimmerman noted that elevated ALT accompanied by jaundice was associated with a mortality between 5% and 50%. This observation has since been reffered as Hy’s rule and is currently employed by FDA in evaluation of hepatotoxicity for newly developed drugs.
- Liver injury is defined as an alanine aminotransferase (ALT) level of more than three times the upper limit of the normal range, an alkaline phosphatase (ALP) level of more than twice the upper limit of normal, or a total bilirubin (TBL) level of more than twice the upper limit of normal, associated with any elevation aminotransferases.
- No other reason can be found to explain the combination of increased AT and TBL, such as viral hepatitis A, B, or C; preexisting or acute liver disease; or another drug capable of causing the observed injury .
Note: Elevations in serum enzyme levels are taken as indicators of liver injury, whereas increases in both total and conjugated bilirubin levels, (additionally INR , Serum albumin) are measures of overall liver function.
Serum-chemistry tests must be supplemented by additional clinical evidence ( a detailed history, consideration to individual risk factors, imaging studies, detail medication history including OTC, dietary supplements, herbal medications) to determine accurately whether the injury has been caused by disease or a drug. Of all , most importantly a HIGH INDEX OF SUSPICION in patients presenting with jaundice or abnormal LFTs might avoid significant mobidity and mortality due to DILI.
Clinical scales available for diagnosis of DILI - causality assessment scales
These scales have been utilised in the assessment of DILI risk in clinical trials, post marketing surveillance to assess the causality relationship between drug exposure and development of DILI. The following figure depicts a few of them-

Digestive Disease Week Japan (DDW-J), Maria and Victorino (M&V)- clinical diagnostic scale (CDS), The Naranjo Adverse Drug Reactions Probability Scale (NADRPS), Council for International Organizations of Medical Sciences (CIOMS) or Roussel Uclaf Causality Assessment Method (RUCAM).
Treatment:
No specific treatment is indicated for drug-induced hepatic disease. Treatment is largely supportive and based on symptomatology. The main treatment for DILI is withdrawal of the offending drug. Early recognition of drug toxicity is important to permit assessment of severity and monitoring for acute liver failure. Specific therapy against drug-induced liver injury is limited to the use of N -acetylcysteine in the early phases of acetaminophen toxicity. L-carnitine is potentially valuable in cases of valproate toxicity. In general, corticosteroids have no definitive role in treatment. They may suppress the systemic features associated with hypersensitivity or allergic reactions. Management of protracted drug-induced cholestasis is similar to that for primary biliary cirrhosis. Cholestyramine may be used for alleviation of pruritus. Ursodeoxycholic acid may be used. Lastly, second opinion from the hepatologist may be saught. The development of jaundice following introduction of a drug potentially portends a poor prognosis and should also prompt immediate referral to a center with expertise in hepatology.
Rechallenge: Generally, rechallenge of subjects with significant AT elevations (>5xULN) should not be attempted. If such subjects are rechallenged, they should be followed closely. Rechallenge can be considered if the subject has shown important benefit from the drug and other options are not available or if substantial accumulated data with the test drug do not show a potential for severe injury. The subject should be made aware of the potential risk, and consent to the rechallenge, and the institutional review board consulted.
Prevention:
Predicting the ADR is not an easy task. since animal models do not adequtely represent DILI in preclinical testing and also given the average no patients (max 3000) registered in phase 3 , clinical trials , under predicts the true risk of drug’s hepatotoxic potential. Perhaps the best time to recognize DILI is post marketing surveillance.
1. Surveillance programmes such as reporting ADR to FDA at MedWatch or by calling 1-800-FDA-1088.
2. The DILIN network : The Drug-Induced Liver Injury Network (DILIN) ( http://dilin.dcri.duke.edu ) was established in 2003 with the aim of studying the problem of hepatotoxicity. Supported by the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) and the National Institutes of Health. . The goals of this network include creating a registry of documented DILI cases and a bank of biological specimens, which will hopefully facilitate the identification of clinical, immunological, and environmental risk factors of DILI.
3. Keeping an eye on case reports that appear in medical literature to help guide or confirm suspicion of DILI.
4. Educating the patient on recognition of symptoms of DILI and adviced to promptly seek medical advice.
Recently withdrawn drugs from the market:
Troglitazone, bromfenac,,nimesulide,,ebrotidine,nefazodone,ximelagatran,kava kava, pemoline
Other drugs that have significant limitations of use because of their hepatotoxic effects are felbamate (Felbatol), an antiepileptic used for complex partial seizures; zileuton (Zyflo), indicated for asthma; tolcapone (Tasmar), used for Parkinson disease; trovafloxacin (Trovan), an antibiotic; benoxaprofen, an NSAID; and tienilic acid, a diuretic.
Recent warnings issued by the FDA:
- Propylthiouracil - indicated for hyperthyroidism due to Graves disease.
- Interferon beta-1a (Avonex) - used in treatment of multiple sclerosis.
- Telithromycin is an antibiotic of the ketolide class, approved by the FDA in April 2004 for the treatment of respiratory infections in adults.
- Duloxetine (an anti-depressant)
Conclusion:
Exploitation of the growing body of knowledge of genetic polymorphisms, through the field of pharmacogenomics, should revolutionize our ability to prevent hepatotoxicity. The emerging fields of proteomics and metabonomics also seem to promise insights into the mechanisms of drug-related hepatotoxicity.
0 comments